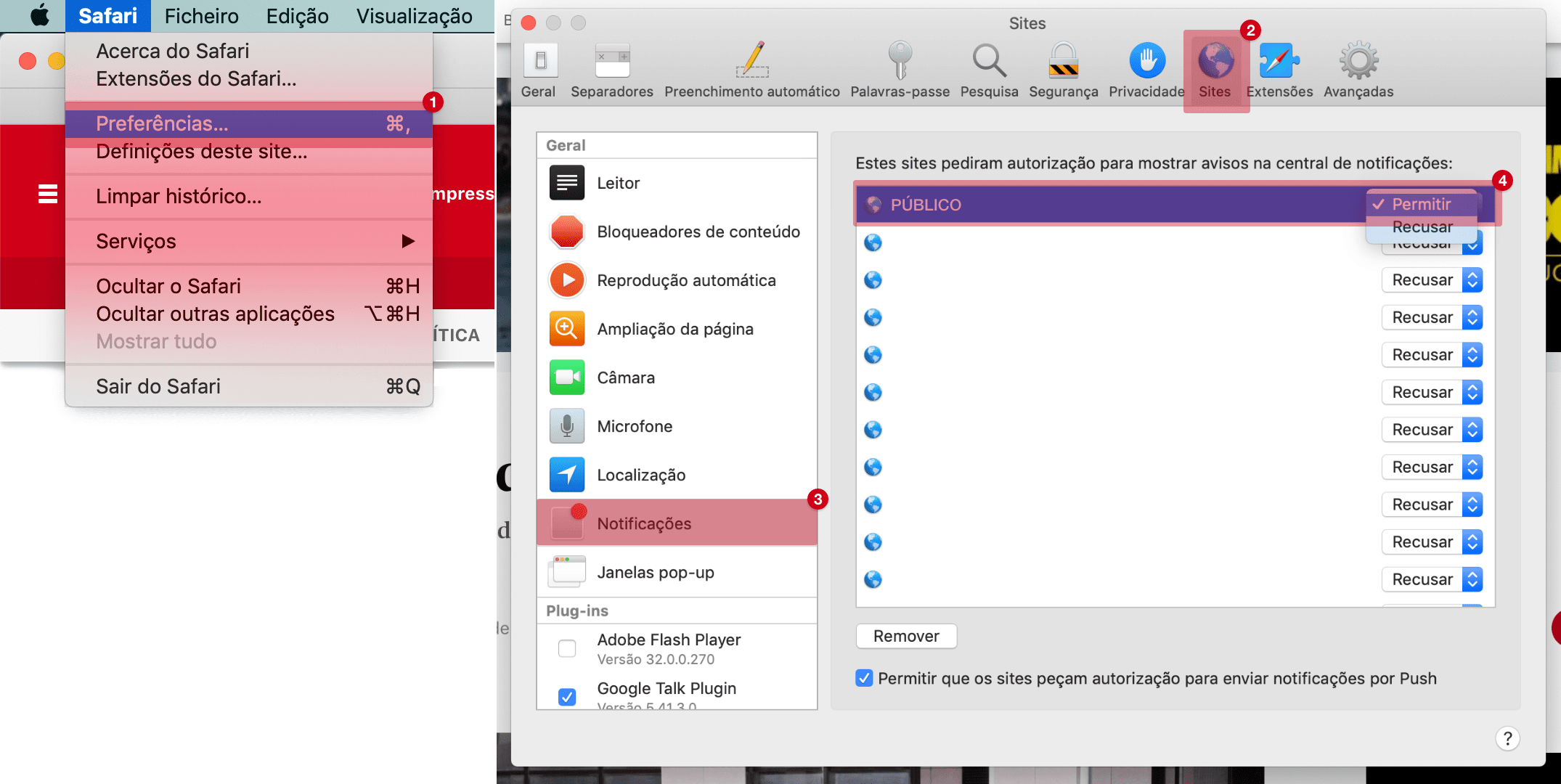
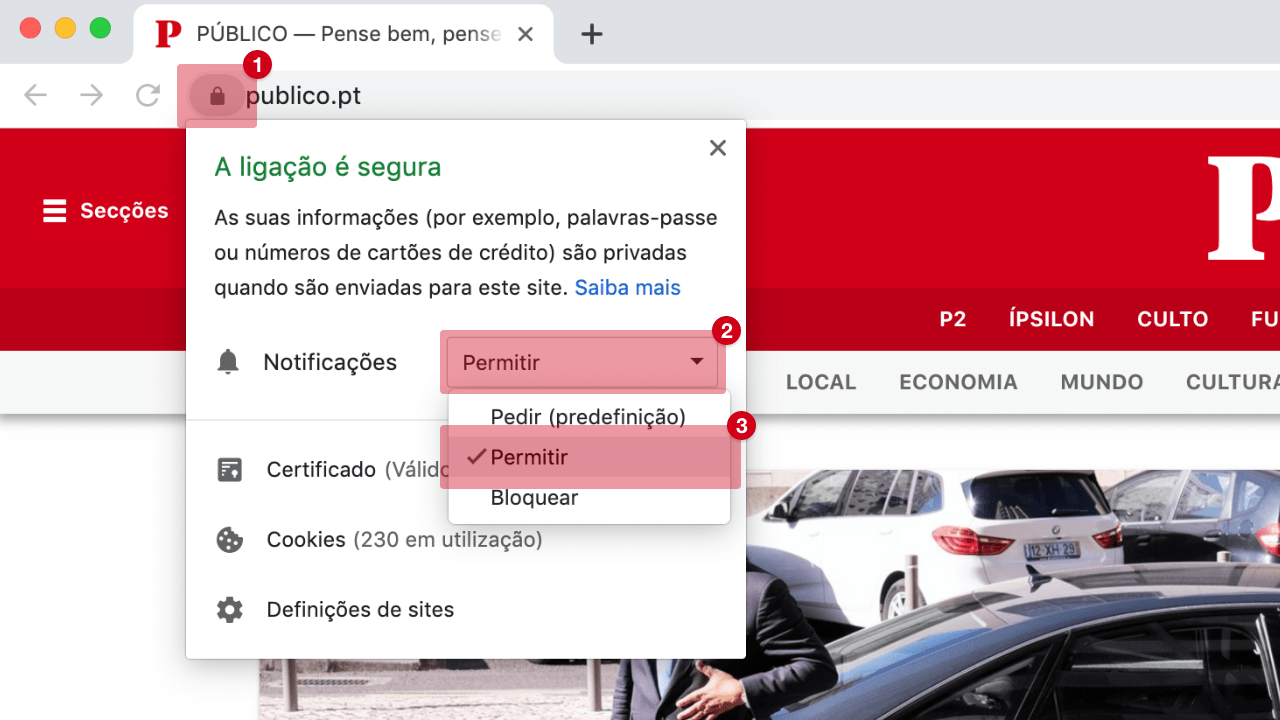
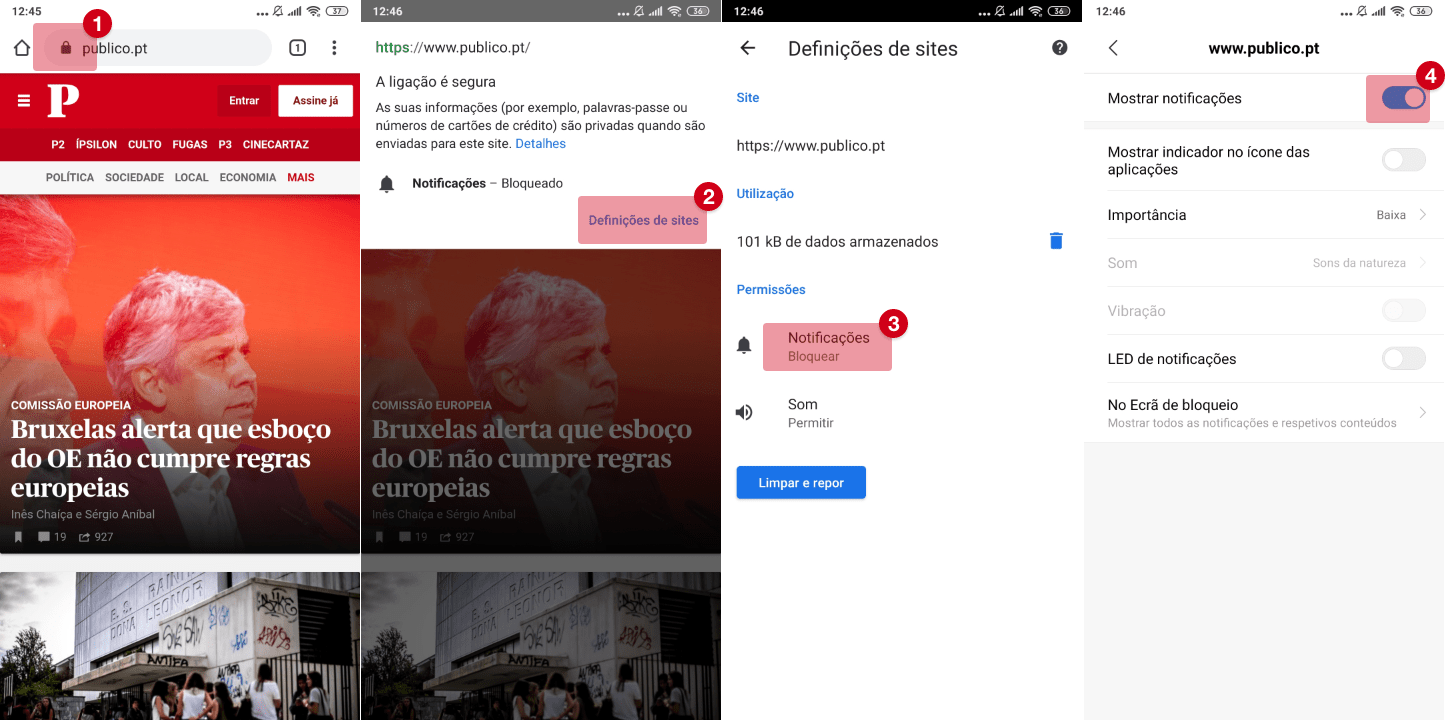
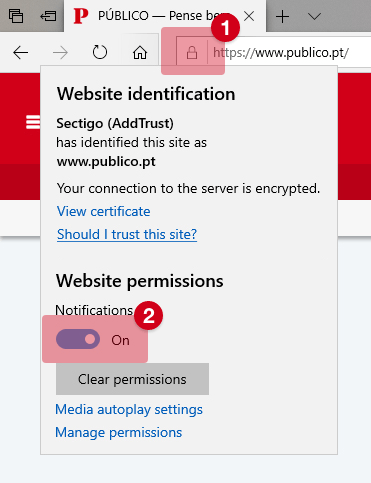
Covid-19: Agência Europeia de Medicamentos começou a analisar a vacina russa Sputnik V
Regulador europeu decidiu iniciar este exame em tempo real com base em estudos com resultados positivos já efectuados.

A Agência Europeia de Medicamentos (EMA) anunciou esta quinta-feira que iniciou uma “análise contínua” da vacina russa contra a covid-19, a Sputnik V, para determinar a sua conformidade com os requisitos da UE em matéria de eficácia, segurança e qualidade.
Em comunicado, o regulador europeu anuncia que decidiu iniciar este exame em tempo real “com base nos resultados de estudos laboratoriais e estudos clínicos em adultos” já realizados, que “indicam que a Sputnik V desencadeia a produção de anticorpos e células imunitárias que visam o coronavírus SARS-CoV-2 e que podem ajudar a proteger contra a covid-19”.
“A EMA avaliará os dados à medida que estes se tornem disponíveis para decidir se os benefícios superam os riscos. A revisão contínua prosseguirá até estarem disponíveis provas suficientes para um pedido formal de autorização de comercialização [desta vacina]”, explica a agência.
A agência “avaliará a conformidade da Sputnik V com os requisitos habituais da UE em termos de eficácia, segurança e qualidade” e, ressalvando que não pode prever prazos para uma decisão, sublinha que um parecer sobre um eventual pedido de comercialização “deverá levar menos tempo do que o normal”, devido ao trabalho realizado durante esta revisão contínua.
“A EMA comunicará mais tarde, quando o pedido de autorização de comercialização da vacina tiver sido apresentado”, adianta a agência.
A chamada “revisão contínua” é um instrumento regulador que a EMA utiliza para acelerar a avaliação de um medicamento promissor durante uma emergência de saúde pública, já que, ao rever os dados em tempo real, à medida que estes ficam disponíveis, pode chegar mais cedo a um parecer final sobre a autorização de comercialização, quando esta der entrada.
Regra geral, todos os dados sobre a eficácia, segurança e qualidade de um medicamento ou vacina e todos os documentos necessários devem estar prontos no início da avaliação num pedido formal de autorização de introdução no mercado.
No caso de uma avaliação contínua, o Comité de Medicamentos para Uso Humano da EMA revê os dados à medida que estes ficam disponíveis a partir de estudos em curso, e, quando este comité decidir que existem dados suficientes disponíveis, a empresa pode apresentar um pedido formal de aprovação.
Até ao momento, a EMA deu luz verde a três vacinas para a covid-19: a da Pfizer-BioNTech (também conhecida como “Comirnaty”), a 21 de Dezembro de 2020, a da Moderna, a 6 de Janeiro, e da AstraZeneca, em 29 de Janeiro, e deverá tomar na próxima semana uma decisão sobre a vacina da Johnson & Johnson, produzida pela farmacêutica Janssen.